400-600-8767
400-600-8767 export@labonce.com
export@labonce.com ch
ch en
en










|
STABILITY TESTING: PHOTOSTABILITY TESTING OF NEW PHARMACEUTICAL SUBSTANCES AND PRODUCTS
|
|
1. GENERAL
The ICH Harmonized Tripartite Guideline covering the Stability Testing of New Pharmaceutical Substances and Products (hereafter referred to as the Parent Guideline) notes that light testing should be an integral part of stress testing. This document is an annex to the Parent Guideline and addresses the recommendations for photostability testing.
A. Preamble
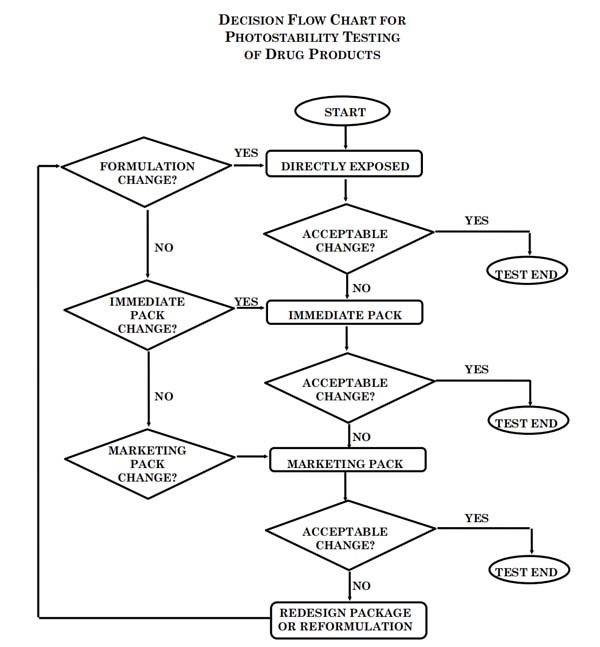
The intrinsic photostability characteristics of new pharmaceutical substances and products should be evaluated to demonstrate that, as appropriate, light exposure does not result in unacceptable change. Normally, photostability testing is carried out on a single batch of material selected as described under Selection of Batches in the Parent Guideline. Under some circumstances these studies should be repeated if certain variations and changes are made to the product (e.g., formulation, packaging).Whether these studies should be repeated depends on the photostability characteristics determined at the time of initial filing and the type of variation and/orchange made. The guideline primarily addresses the generation of photostability information for submission in Registration Applications for new molecular entities and associated pharmaceutical products. The guideline does not cover the photostability of pharmaceutical after administration (i.e. under conditions of use) and those applications not covered by the Parent Guideline. Alternative approaches may be used if they are scientifically sound and justification is provided. A systematic approach to photostability testing is recommended covering, as appropriate, studies such as: i) Tests on the pharmaceutical substance; ii) Tests on the exposed pharmaceutical product outside of the immediate pack;and if necessary ; iii) Tests on the pharmaceutical product in the immediate pack; and if necessary ; iv) Tests on the pharmaceutical product in the marketing pack. The extent of drug product testing should be established by assessing whether or not acceptable change has occurred at the end of the light exposure testing as described in the Decision Flow Chart for Photostability Testing of Pharmaceutical Products. Acceptable change is change within limits justified by the applicant. The formal labeling requirements for photolabile pharmaceutical substances and drug products are established by national/regional requirements.
1 Photostability Testing of New pharmaceutical Substances and Products
B. Light Sources
The light sources described below may be used for photostability testing. The applicant should either maintain an appropriate control of temperature to minimize the effect of localized temperature changes or include a dark control in the same environment unless otherwise justified. For both options 1 and 2, a pharmaceutical manufacturer/applicant may rely on the spectral distribution specification of the light source manufacturer.
Option 1
Any light source that is designed to produce an output similar to the D65/ID65 emission standard such as an artificial daylight fluorescent lamp combining visible and ultraviolet (UV) outputs, xenon, or metal halide lamp. D65 is the internationally recognized standard for outdoor daylight as defined in ISO 10977 (1993). ID65 is the equivalent indoor indirect daylight standard. For a light source emitting significant radiation below 320 nm, an appropriate filter(s) may be fitted to eliminate such radiation.
Option 2
For option 2 the same sample should be exposed to both the cool white fluorescent and near ultraviolet lamp. 1. A cool white fluorescent lamp designed to produce an output similar to that specified in ISO 10977(1993) ; 2. A near UV fluorescent lamp having a spectral distribution from 320 nm to 400 nm with a maximum energy emission between 350 nm and 370 nm; a significant proportion of UV should be in both bands of 320 to 360 nm and 360 to 400 nm.
C. Procedure
For confirmatory studies, samples should be exposed to light providing an overall illumination of not less than 1.2 million lux hours and an integrated near ultraviolet energy of not less than 200 watt hours/square meter to allow direct comparisons to be made between the pharmaceutical substance and drug product. Samples may be exposed side-by-side with a validated chemical actinometric system to ensure the specified light exposure is obtained, or for the appropriate duration of time when conditions have been monitored using calibrated radiometers/lux meters. An example of an actinometric procedure is provided in the Annex. If protected samples (e.g., wrapped in aluminum foil) are used as dark controls to evaluate the contribution of thermally induced change to the total observed change,these should be placed alongside the authentic sample.
2. PHARMACEUTICAL SUBSTANCE
For pharmaceutical substances, photostability testing should consist of two parts: forced degradation testing and confirmatory testing.
The purpose of forced degradation testing studies is to evaluate the overall
photosensitivity of the material for method development purposes and/or degradation pathway elucidation. This testing may involve the pharmaceutical substance alone and/or in simple solutions/suspensions to validate the analytical procedures. In these studies, the samples should be in chemically inert and transparent containers. In these forced degradation studies, a variety of exposure conditions may be used, depending on the photosensitivity of the pharmaceutical substance involved and the intensity of the light sources used. For development and validation purposes it is appropriate to limit exposure and end the studies if extensive decomposition occurs. For photostable materials, studies may be terminated after an appropriate exposure level has been used. The design of these experiments is left to the applicant’s discretion although the exposurelevels used should be justified. Under forcing conditions, decomposition products may be observed that are unlikely to be formed under the conditions used for confirmatory studies. This information may be useful in developing and validating suitable analytical methods. If in practice it has been demonstrated they are not formed in the confirmatory studies, these degradation products need not be further examined.
Confirmatory studies should then be undertaken to provide the information necessary
for handling, packaging, and labeling (see section I.C., Procedure, and II.A.,
Presentation, for information on the design of these studies).
Normally, only one batch of drug substance is tested during the development phase,and then the photostability characteristics should be confirmed on a single batch selected as described in the Parent Guideline if the pharmaceutical is clearly photostable or photolabile. If the results of the confirmatory study are equivocal, testing of up to two additional batches should be conducted. Samples should be selected as described in the Parent Guideline.
A. Presentation of Samples
Care should be taken to ensure that the physical characteristics of the samples under test are taken into account and efforts should be made, such as cooling and/or placing the samples in sealed containers, to ensure that the effects of the changes in physical states such as sublimation, evaporation or melting are minimized. All such precautions should be chosen to provide minimal interference with the exposure of samples under test. Possible interactions between the samples and any material used for containers or for general protection of the sample, should also be considered and eliminated wherever not relevant to the test being carried out. As a direct challenge for samples of solid pharmaceutical substances, an appropriate amount of sample should be taken and placed in a suitable glass or plastic dish and protected with a suitable transparent cover if considered necessary. Solid pharmaceutical substances should be spread across the container to give a thickness of typically not more than 3 millimeters.Pharmaceutical substances that are liquids should be exposed in chemically inert and transparent containers.
4
Photostability Testing of New Pharmaceutical Substances and Products
B. Analysis of Samples
At the end of the exposure period, the samples should be examined for any changes in physical properties (e.g., appearance, clarity, or color of solution) and for assay and degradants by a method suitably validated for products likely to arise from photochemical degradation processes. Where solid Pharmaceutical substance samples are involved, sampling should ensure that a representative portion is used in individual tests. Similar sampling considerations,such as homogenization of the entire sample, apply to other materials that may not be homogeneous after exposure. The analysis of the exposed sample should be performed concomitantly with that of any protected samples used as dark controls if these are used in the test.
C. Judgement of Results
The forced degradation studies should be designed to provide suitable information to develop and validate test methods for the confirmatory studies. These test methods should be capable of resolving and detecting photolytic degradants that appear during the confirmatory studies. When evaluating the results of these studies, it is important to recognize that they form part of the stress testing and are not therefore designed to establish qualitative or quantitative limits for change. The confirmatory studies should identify precautionary measures needed in manufacturing or in formulation of the pharmaceutical product, and if light resistant packaging is needed. When evaluating the results of confirmatory studies to determine whether change due to exposure to light is acceptable, it is important to consider the results from other formal stability studies in order to assure that the pharmaceutical will be within justified limits at time of use (see the relevant ICH Stability and Impurity Guidelines).
3. PHARMACEUTICAL PRODUCT
Normally, the studies on pharmaceutical products should be carried out in a sequential manner starting with testing the fully exposed product then progressing as necessary to the product in the immediate pack and then in the marketing pack. Testing should progress until the results demonstrate that the pharmaceutical product is adequately protected from exposure to light. The pharmaceutical product should be exposed to the light conditions described under the procedure in section I.C. Normally, only one batch of pharmaceutical product is tested during the development phase, and then the photostability characteristics should be confirmed on a single batch selected as described in the Parent Guideline if the product is clearly photostable or photolabile. If the results of the confirmatory study are equivocal, testing of up to two additional batches should be conducted. For some products where it has been demonstrated that the immediate pack is completely impenetrable to light, such as aluminium tubes or cans, testing should normally only be conducted on directly exposed drug product. It may be appropriate to test certain products such as infusion liquids, dermal creams, etc., to support their photostability in-use. The extent of this testing should depend on and relate to the directions for use, and is left to the applicant’s discretion.
The analytical procedures used should be suitably validated.
5 Photostability Testing of New Pharmaceutical Substances and Products
A. Presentation of Samples
Care should be taken to ensure that the physical characteristics of the samples under test are taken into account and efforts, such as cooling and/or placing the samples in sealed containers, should be made to ensure that the effects of the changes in physical states are minimized, such as sublimation, evaporation, or melting. All such precautions should be chosen to provide a minimal interference with the irradiation of samples under test. Possible interactions between the samples and any material used for containers or for general protection of the sample should also be considered and eliminated wherever not relevant to the test being carried out. Where practicable when testing samples of the drug product outside of the primary pack, these should be presented in a way similar to the conditions mentioned for the pharmaceutical substance. The samples should be positioned to provide maximum area of exposure to the light source. For example, tablets, capsules, etc., should be spread in a single layer. If direct exposure is not practical (e.g., due to oxidation of a product), the sample should be placed in a suitable protective inert transparent container (e.g., quartz). If testing of the drug product in the immediate container or as marketed is needed,the samples should be placed horizontally or transversely with respect to the light source, whichever provides for the most uniform exposure of the samples. Some adjustment of testing conditions may have to be made when testing large volume containers (e.g., dispensing packs).
B. Analysis of Samples
At the end of the exposure period, the samples should be examined for any changes in physical properties(e.g.,appearance,clarity or color of solution,dissolution/disintegration for dosage forms such as capsules, etc.) and for assay and degradants by a method suitably validated for products likely to arise from photochemical degradation processes.
When powder samples are involved, sampling should ensure that a representative portion is used in individual tests. For solid oral dosage form products, testing should be conducted on an appropriately sized composite of, for example, 20 tablets or capsules. Similar sampling considerations, such as homogenization or solubilization of the entire sample, apply to other materials that may not be homogeneous after exposure (e.g., creams, ointments, suspensions, etc.). The analysis of the exposed sample should be performed concomitantly with that of any protected samples used as dark controls if these are used in the test.
C. Judgement of Results
Depending on the extent of change special labeling or packaging may be needed to mitigate exposure to light. When evaluating the results of photostability studies to determine whether change due to exposure to light is acceptable, it is important to consider the results obtained from other formal stability studies in order to assure that the product will be within proposed specifications during the shelf life (see the relevant ICH Stability and Impurity Guidelines).
6 Photostability Testing of New Pharmaceutical Substances and Products
4. ANNEX
A. Quinine Chemical Actinometry
The following provides details of an actinometric procedure for monitoring exposure toa near UV fluorescent lamp (based on FDA/National Institute of Standards and Technology study). For other light sources/actinometric systems, the same approachmay be used, but each actinometric system should be calibrated for the light sourceused. Prepare a sufficient quantity of a 2 per cent weight/volume aqueous solution of quinine monohydrochloride dihydrate (if necessary, dissolve by heating).
Option 1
Put 10 milliliters (ml) of the solution into a 20 ml colorless ampoule seal it hermetically, and use this as the sample. Separately, put 10 ml of the solution into a 20 ml colourless ampoule (see note 1), seal it hermetically, wrap in aluminum foil to protect completely from light, and use this as the control. Expose the sample and control to the light source for an appropriate number of hours. After exposure determine the absorbances of the sample (AT) and the control (Ao) at 400 nm using a
1 centimeter (cm) path length. Calculate the change in absorbance, Δ A = AT - Ao. The length of exposure should be sufficient to ensure a change in absorbance of at least 0.9.
Option 2
Fill a 1 cm quartz cell and use this as the sample. Separately fill a 1 cm quartz cell,wrap in aluminum foil to protect completely from light, and use this as the control. Expose the sample and control to the light source for an appropriate number of hours.
After exposure determine the absorbances of the sample (AT) and the control (Ao) at 400 nm. Calculate the change in absorbance, Δ A = AT - Ao. The length of exposure should be sufficient to ensure a change in absorbance of at least 0.5. Alternative packaging configurations may be used if appropriately validated. Alternative validated chemical actinometers may be used.
Note 1: Shape and Dimensions (See Japanese Industry Standard (JIS) R3512 (1974) for ampoule specifications)
Photostability Testing of New Pharmaceutical Substances and Products
5. GLOSSARY
Immediate (primary) pack is that constituent of the packaging that is in direct contact with the drug substance or pharmaceutical product, and includes any appropriate label. Marketing pack is the combination of immediate pack and other secondary packaging such as a carton. Forced degradation testing studies are those undertaken to degrade the sample deliberately. These studies, which may be undertaken in the development phase normally on the pharmaceutical substances, are used to evaluate the overall photosensitivity of the material for method development purposes and/or degradation pathway elucidation. Confirmatory studies are those undertaken to establish photostability characteristics under standardized conditions. These studies are used to identify precautionary measures needed in manufacturing or formulation and whether light resistant packaging and/or special labeling is needed to mitigate exposure to light. For the confirmatory studies, the batch(es) should be selected according to batch selection for long-term and accelerated testings which is described in the Parent Guideline.
6. REFERENCES
Quinine Actinometry as a method for calibrating ultraviolet radiation intensity in light-stability testing of pharmaceuticals. Yoshioka S. et al., Pharmaceutical Development and Industrial Pharmacy, 20 (13), 2049 - 2062(1994).
|